Premier PSM Research
 Muhammad (MO) Butt
Muhammad (MO) Butt
Research Coordinator Dr. Sanober Kable
Dr. Sanober Kable
Principal Investigator Theresa Englutt
Theresa Englutt
Sub Investigator
The Premier PSM Research Department supports cutting-edge research in Pulmonary Related Diseases and Medicine, offering numorious studies for Asthma, COPD, IPF, PAH and many more at dedicated research centers. Several research groups report directly to the Principle Investigator (PI), Dr. Sanober Kable, most research studies are based in individual clinical settings, where they engage in interdisciplinary research that complements the academic goals of their departments.
- Gender: All
- Age: 18 Years and over
- Phase: Observational
- IRB Number: SSU00221999
- Healthy Volunteers: This study is accepting healthy volunteers
- Males and females at least 18 years of age who give voluntary written informed consent to participate in the study.
- Patients with a diagnosis of ILD based on computed tomography
imaging, including:
- Idiopathic interstitial pneumonia, including idiopathic pulmonary fibrosis
- Connective tissue disease-associated ILD with FVC <70%
- Hypersensitivity pneumonitis
- Scleroderma-related ILD
- Autoimmune ILD
- Nonspecific interstitial pneumonia
- Occupational lung disease
- Combined pulmonary fibrosis and emphysema
- Patients must meet a total of 2 or more criteria from at least 2 distinct categories below (ie, Category 1, Category 2, Category 3) based on the Investigator’s clinical judgment, within 180 days of Screening.
- DLCO <40%
- DLCO decline of ≥15% based on 2 most recent assessments
- Worsening DLCO (decline >10%) with stable FVC (decline <5%) (ie, worsening FVC/DLCO ratio) based on 2 most recent assessments
- Right ventricle enlargement: RV:LV ratio >1
- Pulmonary artery enlargement
- Pulmonary artery/aorta ratio >1.0
- Ventricular septal flattening
- Enlarged pulmonary arteries in the lung periphery
- Symptoms disproportionate to ILD severity or changes in symptoms not explained by ILD progression
- Desaturation on 6MWT disproportionate to ILD severity in the opinion of the Investigator
- 6-Minute Walk Distance decline of ≥15% based on 2 more recent assessments
- Worsening desaturation requiring more supplemental oxygen
- Recent worsening desaturation
- BNP >200 pg/mL or NT-proBNP >395 pg/mL
- Physical exam findings (at least 1 of the following): syncope, jugular venous distension, ankle swelling/peripheral edema, ascites, loud P2 or S2 heart sound, or hepatomegaly
- RV systolic pressure >35 mmHg
- Any RV dilation or enlargement
- Other RV abnormalities in the clinician’s judgment
- Tricuspid annular plane systolic excursion <2 cm
Category 1: Clinical tests
a. PFTs with at least 1 of the following results:
b. Historical HRCT with any of the following findings:
Category 2: Symptoms, oxygenation, exercise capacity, and BNP/NT-proBNP
Category 3: Echocardiography
Patients are excluded from the study if any of the following criteria apply:
- Prior RHC with mPAP >20 mmHg
- Currently on a Food and Drug Administration (FDA)-approved pulmonary arterial hypertension medication
- Diagnosed with chronic obstructive pulmonary disease
- Uncontrolled or untreated sleep apnea
- Pulmonary embolism within the past 3 months
- History of ischemic heart disease or left-sided myocardial dysfunction within 12 months of Screening, defined as LV ejection fraction <40% or pulmonary capillary wedge pressure>15 mmHg
- Any other clinical features that, in the opinion of the Investigator, might adversely affect interpretation of study data or study safety, or make the patient unsuitable for RHC
- Gender: All
- Age: 18 Years and over
- Phase: Phase 3
- IRB Number: 20210052
- Healthy Volunteers: This study is accepting healthy volunteers
- Age: ≥ 40 years old
- FVC ≥ 45% and ≤ 80% predicted
- If on background pirfenidone or nintedanib for IPF, stable dose for ≥ 30 days prior to Baseline
- Concomitant use of both pirfenidone and nintedanib is not permitted
- Diagnosis of IPF based on the 2018 ATS/ERS/JRS/JRS/ALAT Clinical Practice Guidelines (Raghu 2018)
- HRCT within the last 12 months “consistent with UIP” (confirmed by central reader)
- HCRT may be performed during screening window as study assessment if historic scan within 12 months not available
- Females of child-bearing potential: must have confirmed negative pregnancy test at Screening and Baseline and willing to practice abstinence or use 2 forms of birth control for duration of study and 30 days after discontinuing study drug
- Hormonal/IUD and barrier with spermicide
- Males with a partner of childbearing potential: must use a condom for the duration of treatment and for at least 48 hours after discontinuing study drug
- Able to communicate effectively with study personnel, and is considered reliable, willing, and likely to be cooperative with protocol requirements
- Pregnant or lactating
- Primary obstructive disease: pre-bronchodilator FEV1/FVC <0.70
- Diagnosis of connective tissue disease-ILD or combined pulmonary fibrosis and emphysema
- >10L/min of supplemental oxygen at rest at Baseline
- Receipt of any prostacyclin therapy, IP receptor agonist, ERA, or PDE5-I or sGC stimulator within 60 days prior to Baseline (washout permitted)
- Intermittent use of PDE5-1 for ED allowed
- Shown intolerance or significant lack of efficacy to a prostacyclin or prostacyclin analogue that resulted in discontinuation or inability to effectively titrate therapy
- Myocardial infarction within 6 months prior to Baseline or unstable angina within 30 days prior to Baseline
- Use of any of the following medications:
- Azathioprine (AZA), cyclosporine, mycophenolate mofetil, tacroliumus, oral corticosteroids (OCS) >20 mg/day or the combination of OCS+AZA+N-acetylcysteine within 30 days prior to Baseline
- Cyclophosphamide within 60 days prior to Baseline
- Rituximab within 6 months prior to Baseline
- Exacerbation of IPF or active pulmonary or upper respiratory infection within 30 days prior to Baseline
- Antibiotic or steroid regimens for infection or acute exacerbation must be completed more than 30 days prior to Baseline
- Must have been discharged more than 90 days prior to Baseline if hospitalized for an acute exacerbation of IPF or a pulmonary or upper respiratory infection
- Any condition that would interfere with the interpretation of study assessments or would impair study participation or cooperation
- Gender: All
- Age: 18 Years and over
- Phase: Phase 3
- IRB Number:SSU00167839
- Healthy Volunteers: This study is accepting healthy volunteers
- Age 18 - 75 y, BMI 19.0 - 32.0 kg/m2 (inclusive)
- WHO Group 1 PH (PAH):
- Right heart catheterization (RHC) at screening or within 30 days of screening with the following hemodynamics:
- PAP ≥ 25 mmHg, PCWP ≤ 15 mmHg, & PVR of ≥ 5 WU
- NYHA/WHO Functional capacity Class II-III
- No evidence of thromboembolic disease
- No more than 2 medications from the following classes: Endothelin receptor antagonists (eg, ambrisentan, bosentan, macitentan), phosphoesterase type 5 inhibitors (eg, sildenafil, tadalafil), guanylate cyclase stimulator (eg, riociguat)
- Stable PH medications for 60 days
- FVC ≥70% within 1 year of Screening
At least two 6MWTs at Screening between 150 - 550 m, both values are w/i 15% of each other
- PH Group 1 as described in IC
- Hypersensitivity / contraindication to TPIP or TRE or mannitol
- Previous intolerance to prostacyclin analogs or receptor agonists (eg, selexipag)
- QTcF interval > 480 ms
- Known ventricular or supraventricular tachyarrhythmia (except for paroxysmal atrial fibrillation), or any symptomatic bradycardia
- LVEF ≤ 40%
- SBP < 90 mmHg
- Renal or liver disease
- HIV or Hep B or C
- Triple therapy of endothelin receptor agonists, phosphoesterase type 5 inhibitors, and guanylate cyclase stimulators
- Gender: All
- Age: 18 Years and over
- Phase: Phase
- IRB Number:20203428
- Healthy Volunteers: This study is accepting healthy volunteers
MDI Use
- Proper administration technique - spacers not permitted
Reproduction
- Pregnancy test to be performed on all women of childbearing potential.
- Acceptable contraceptive measures
Permitted Asthma Meds During Screening
- Run-in BFF MDI BID and albuterol prn only;
- SCS or ICS for the treatment of an asthma exacerbation (defined use)
eDiary Compliance
- 14 day compliance ≥70% during screening
Respiratory Infection During Screening:
- Completed treatment more than 4 weeks prior to randomization (V5)
Asthma Exacerbation During Screening
- Completed treatment with SCS and/or additional ICS more than 4 weeks prior to V5
Asthma History
- Life- threatening asthma
- Hospitalization for asthma w/in 2 months of V1
Nebulizer Usage
- Regular use of a nebulizer or a home nebulizer for receiving asthma medications
Oral B2- agonist Usage
- Use of oral 2-agonist within 3 months of V1
SCS Usage
- Oral and IV CS use (any dose) except for exacerbation within 4 weeks of V1
Immuno-modulators/ immuno-suppressive Usage
- Use of any immunomodulators or immunosuppressive meds within 3 months or 5 half-lives, whichever is longer, and not permitted during study duration
Medical conditions
- Historical or current evidence of a clinically significant disease; clinically relevant abnormal findings on P.E., clinical labs, or ECG
Eye Disorders
- Narrow angle glaucoma not adequately treated and/or change in vision that may be
relevant, in the opinion of the Investigator, w/in 3 months of V1
Smoking history
- Current smokers, former smokers with >10 pack-years hx or who stopped smoking <6 months prior to V1 (includes marijuana, e-cigs & vaping devices)
- Gender: All
- Age: 18 Years and over
- Phase: Phase 3
- IRB Number:SSU00223828
- Healthy Volunteers: This study is NOT accepting healthy volunteers
Study Phases
Clinical trials have four phases, or steps, in the clinical research process. You may notice when you see a name of a clinical trial that it includes Phase I, II, III, or IV, or it may be written as Phase 1, 2, 3, or 4. Here’s what those phases mean:

Phase 1
Researchers test a drug or treatment in a small group of people (20-100) for the first time. The purpose is to study the drug or treatment to learn about safety and identify side effects.

Phase 2
The new drug or treatment is given to a larger group of people (100-300) to determine its effectiveness and to further study its safety.

Phase 3
The new drug or treatment is given to large groups of people (1,000-3,000) to confirm its effectiveness, monitor side effects, compare it with standard or similar treatments, and collect information that will allow the new drug or treatment to be used safely.

Phase 4
After a drug is approved by the FDA and made available to the public, researchers track its safety in the general population, seeking more information about a drug or treatment's benefits, and optimal use.
What Can I Expect?
Here’s what happens before and after you enroll in a clinical study or clinical trial.
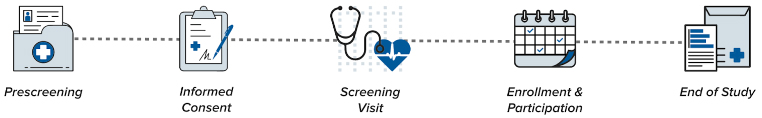
Prescreening
For trials enrolling patients with a particular medical condition, some pre-screening may happen behind-the-scenes before we meet with you. For studies enrolling healthy volunteers, staff members explain the trial in detail and gather more information about you during the pre screening process.
Informed Consent
Informed consent is an essential part of participating in a clinical study. It is the process of learning the key facts about a clinical study before deciding whether to participate. Once you have had all your questions answered, and if you agree to participate, you may be asked to sign an informed consent form. Participants should take the time to review the informed consent document carefully and decide if they feel comfortable with participating in the study.
You will get a copy of the document, which you can keep to refer back to in the future. Also, note that informed consent is a continuous process that does not end with a signed document. You should always feel free to ask questions about your participation in a study at any time during or after your participation ends. The researchers will also provide you any new information during the study if it could affect your willingness to participate.
Screening Visit
Once you have consented to participate in a study, you may be asked to undergo other procedures and tests, such as filling out questionnaires or having blood work, to confirm that you qualify for the study. You may be asked to make a special visit for screening.
Enrollment and Participation
Once you have enrolled, the study team will review the study procedures with you and schedule tests and other appointments. You will follow the trial procedures and report any issues or concerns to the study team. Remember, participating in a clinical study is totally voluntary and you can decide to stop at any time. Study participants continue to see their regular physician for usual health care throughout the study.
End of Study
Your participation in the study is complete. Researchers may provide participants with information about how they may find results once the study data is analyzed.
How to Sign up...
- Mention the study you are interested in to Dr.Kable for an evaluation
- Please contact MO at 903-465-5012 Ext:110 for further information regarding taking part in our research department.